R.J. Sension T.P. McClain; L.B. Michocki, N.A. Miller, R. Alonso-Mori, F.A. Lima, F. Ardana-Lamas, M. Biednov, T. Chung, A. Deb, Y. Jiang, A.K. Kaneshiro, D. Khakhulin, K. J. Kubarych, R.M. Lamb, J.H. Meadows, F. Otte, D.L. Sofferman, S. Song, Y. Uemura, T.B. van Driel, J.E. Penner-Hahn, J. Phys. Chem. B 128 (2024) 8131–8144
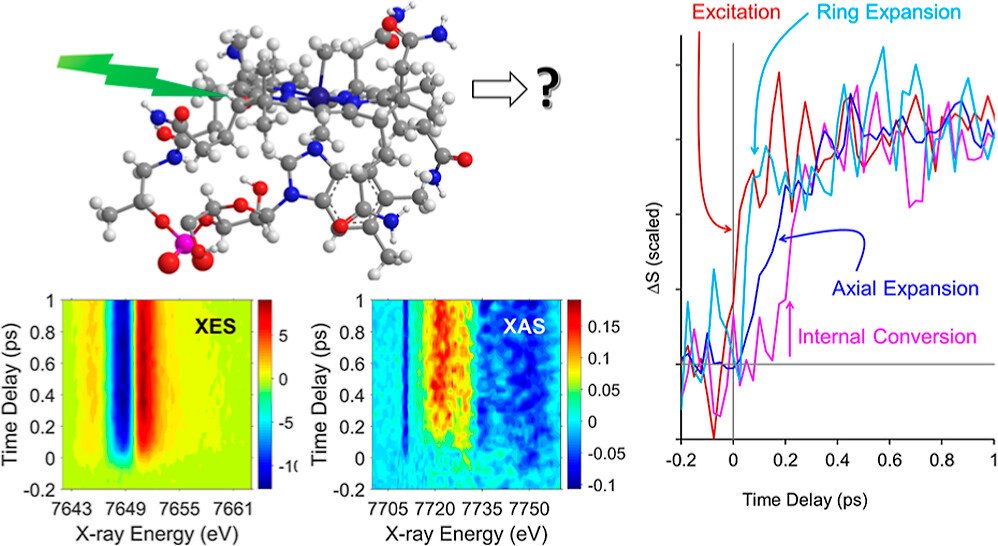
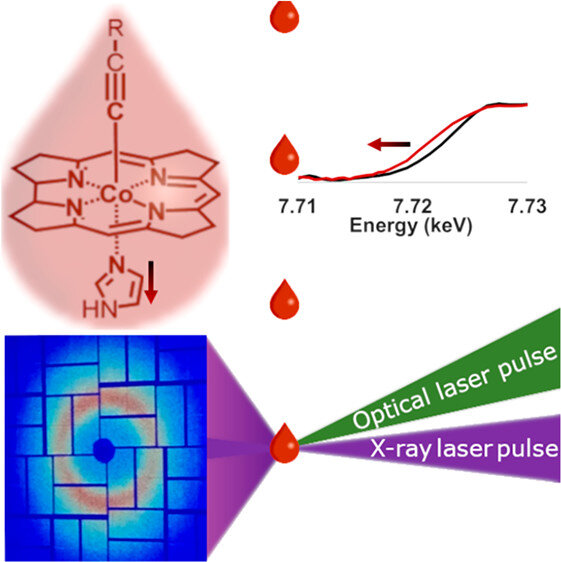
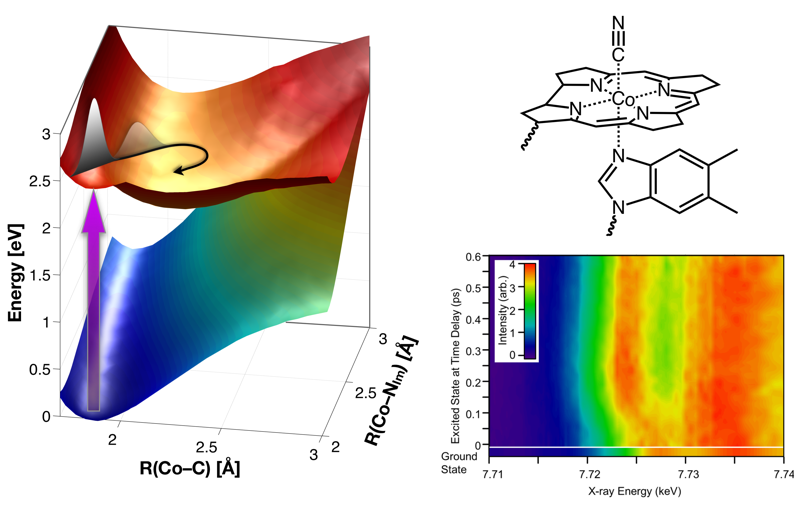
CarH is a protein photoreceptor that uses a form of B12, adenosylcobalamin (AdoCbl), to sense light via formation of a metastable excited state. Aside from AdoCbl bound to CarH, methylcobalamin (MeCbl) is the only other example─to date─of photoexcited cobalamins forming metastable excited states with lifetimes of nanoseconds or longer. The UV–visible spectra of the excited states of MeCbl and AdoCbl bound to CarH are similar. We have used transient Co K-edge X-ray absorption and X-ray emission spectroscopies in conjunction with transient absorption spectroscopy in the UV–visible region to characterize the excited states of MeCbl. These data show that the metastable excited state of MeCbl has a slightly expanded corrin ring and increased electron density on the cobalt, but only small changes in the axial bond lengths.